A New Potent Photooxidant
Pushing the limits of LED driven visible-light photocatalysis requires some creative thinking to get more redox potential out of the tools that are readily available to us. A recent report by Wickens and coworkers demonstrates a unique photochemical approach turning one of the most reducing photocatalysts available into a strong oxidant (Ref 1). And in doing so, enabling the oxidation of some otherwise difficult-to-oxidize compounds.
Using visible-light photocatalysis with a conventional LED light source limits us to energies ~3.2 eV (@390 nm) – 3.4 eV if you want to stretch the limits of “visible-light” and include down to 365 nm light. However, with energy losses from the initial excited state photocatalyst through relaxation, internal conversion, and intersystem crossing, conventional photocatalysts under visible-light irradiation using 1 photon of light limits us to energy in the -2 to +V vs SCE range. This puts many hydrocarbons (and benzene and chlorobenzene) outside the realm of typical visible-light photocatalysis.
One strategy for overcoming this problem is to utilize more than one photon per catalytic cycle, an approach for strong reductions championed by König and coworkers since 2014 (Ref 2). We wrote about a related review of multiphoton excitation here: https://hepatochem.com/multi-photon-approaches-to-synthetic-photochemistry/ This consecutive photon-induced electron transfer (conPET) uses consecutive excitations (See Figure 1) which starts with an initial excitation to generate a photooxidant (step A), then a reduction with a sacrificial reductant (step B) followed by a second excitation to generate a strong reductant (step C). König‘s initial work involved the reduction of chlorobenzene (step D) while Miyake demonstrated a powerful visible-light catalyzed Birch Reduction (Ref 3). Nicewicz with his Mes-Acr catalyst achieved reductions similar to lithium at -3.36 V vs. SCE (Ref 4).
Figure 1: General conPET mechanism for multiple excitations to generate strong reductants.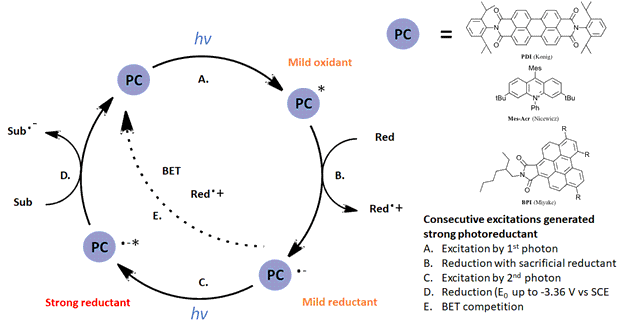
However, before the present work, multiphoton excitations had not been utilized to generate strong oxidants. The authors propose two challenges for this proposed scheme:
- “the catalyst must be a competent photocatalyst in both the closed shell and radical cation states”
- “the terminal oxidant must efficiently activate the catalyst but not otherwise interfere with the reaction”
To start, the authors looked at the oxidative C-N amination of benzene (Eox = 2.5 V vs. SCE) with a pyrazole derivative surveying three photoreductants and six terminal oxidants (Figure 2).
Figure 2: General Scope of C-H Amination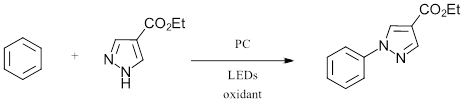
Aminations of this type usually require UV light or strong oxidants such as DDQ. Of the catalysts tested, only N-phenylphenothiazine (PTH) showed significant product formation with O2 selected as the best oxidant (14% yield). The authors then proceeded to switch to a fluorinated solvent (trifluoroethanol-31% yield) and searched for additives to inhibit the superoxide back reaction. With 1 equiv. of LiClO4 as an additive, yield increased to 86%. Interestingly, dropping the loading of LiClO4 to 20 mol% while retaining similar yield (73%) suggests a more complicated role for the lithium additive than simply as a scavenger of superoxide. Adjusting the solvent mixture to a 9:1 TFE:HFIP (hexafluoroisopropanol) solvent results in a 89% yield for the optimized reaction (Figure 3). With the optimized reaction in hand, the authors set out to couple a variety of arenes and pyrazole derivatives (including coupling of pyrazoles containing halogens without dehalogenation) with yields ranging from 45-88%. Check out the full paper for the full substrate scope.
Figure 3: Optimized visible-light oxidative C-N amination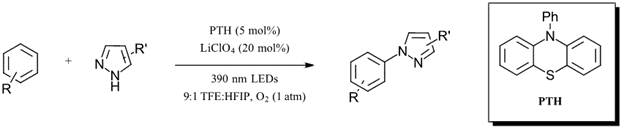
For the multiphoton oxidation as detailed by Wickens and coworkers, the mechanism can be described in Figure 4. As the authors probed the details of their mechanism, a few key points arose. Swapping other organic dyes or Ir(ppy)3 photocatalysts into the optimized reaction resulted in no product formation. While monitoring the time course of the reaction, an induction period was observed with trace product formation followed by zeroth order formation of product until pyrazole is consumed (arene and O2 in excess). If the light is removed, product formation is halted and resumes with return of the light. Additional experiments determined that 2 equiv. of O2 are consumed in the reaction suggesting that O2 is acting as a one-electron oxidant.
Figure 4: Multiphoton oxidation generating a potent photooxidant (modified based on Figure 1 Ref 1)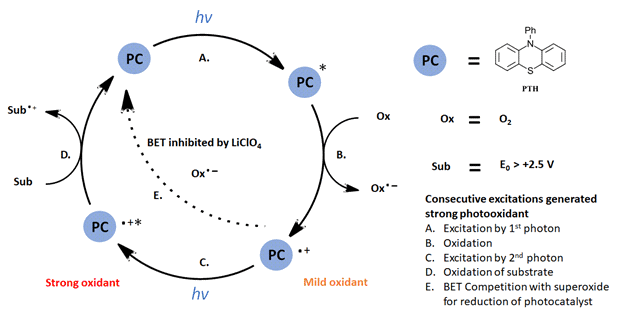
Overall, the system can be described as follows: excitation of the PTH catalyst with 1 photon of 390 nm light (Figure 4-step A); oxidation of PTH* with O2 to generate the PTH radical cation (step 4-B); excitation of the PTH radical cation by a second photon (step 4-C) resulting in an excited catalyst capable of oxidations greater than +2.5 V. Back electron transfer (BET) is inhibited by the presence of a Lewis acid co-catalyst and maintains the catalyst activation. While the mechanism of the reaction may seem complex, operationally the setup is rather simple requiring only commercially available LED light sources and photoreactors (See the Supporting info for an EvoluChem PhotoredoxBox out in the wild). Check out the full paper for a very interesting example of reaction design based on a new mechanism. We would look forward to further expansion on this scheme to other reactions.
A few quick reads for this month:
We’ve been thinking a bit of about photocatalyst design / photocatalyst properties around here lately (more on this to come), so we’ve thought we’d pass a long a good read on organic dye design from the Photochemistry Group at the University of Bologna entitled “Design of BODIPY dyes as triplet photosensitizers: electronic properties tailored for solar energy conversion, photoredox catalysis and photodynamic therapy” (Ref 5).
And finally, while outside the realm of our typical interests in visible-light synthetic chemistry, we would like to make a note of this recent perspective in JACS by Nicholas J. Green, Jianfeng Xu, and John D. Sutherland from Cambridge entitled “Illuminating Life’s Origins: UV Photochemistry in Abiotic Synthesis of Biomolecules”. (Ref 6) An interesting look at all the crazy photochemistry going on during the early days of the Earth.
Check it out in @J_A_C_S – everything you need to know about photochemistry that could have been promoted by the Sun on the young Earth, making the molecules of life!
The main text is more attractive than the TOC, I promise 🙂 #prebioticchemistry #ool
— Nick Green (@NickGreen_chem) April 21, 2021
References:
- Targos, K.; Williams, O. P.; Wickens, Z. K. Unveiling Potent Photooxidation Behavior of Catalytic Photoreductants. Am. Chem. Soc. 2021, 4125–4132. https://doi.org/10.1021/jacs.1c00399.
- Ghosh, I.; Ghosh, T.; Bardagi, J. I.; König, B. “Reduction of aryl halides by consecutive visible light-induced electron transfer processes”. Science 2014, 346, 725-728. https://science.sciencemag.org/content/346/6210/725
- Cole, A. J. P.; Chen, D.; Kudisch, M.; Pearson, R. M.; Miyake, G. M. Organocatalyzed Birch Reduction Driven by Visible Light. Am. Chem. Soc 2020, 142 (31), 13573–13581.
- MacKenzie, I. A.; Wang, L.; Onuska, N. P. R.; Williams, O. F.; Begam, K.; Moran, A. M.; Dunietz, B. D.; Nicewicz, D. A. Discovery and Characterization of an Acridine Radical Photoreductant. Nature 2020, 580 (7801), 76–80. https://doi.org/10.1038/s41586-020-2131-1.
- Bassan, E.; Gualandi, A.; Ceroni, P. Design of BODIPY Dyes as Triplet Photosensitizers”¯: Conversion , Photoredox Catalysis and Photodynamic Therapy. Sci. 2021. ASAP. https://doi.org/10.1039/d1sc00732g.
- Green, N. J.; Xu, J.; Sutherland, J. D. Illuminating Life’s Origins: UV Photochemistry in Abiotic Synthesis of Biomolecules. Am. Chem. Soc 2021. ASAP. https://doi.org/10.1021/jacs.1c01839.
